Japan
サイト内の現在位置


医療機器規制対応と業務効率化の両立をスマートに実現するPLMとは
ものづくりの未来
ER/ES、QMS規制対応と高品質な設計を支援「Obbligato医療機器テンプレート」【2021.06.16】
カテゴリ:設計・開発・技術PLM/CAD
医療機器業界で加速するデジタル化の推進にはFDA Part11をはじめとしたER/ES規制対応が必須です。しかしながらそのハードルは高く、どこから手をつけていいか分からないといった医療機器メーカのご担当者様が数多くいることも事実です。ここでは電子化がなかなか進まない要因や、電子化によるメリットおよび医療機器規制対応を実現するPLM(Product Lifecycle Management)ソリューションについて、事例を交えてご紹介します。
(日本電気株式会社 製造・装置業システム本部主任 澤野陽平)
ER/ESやCSVが成果物の電子化のハードルに

医療機器メーカにおいては、製品開発における成果物が紙での運用という状態がまだまだ続いており、電子化が積年の課題として存在しています。電子化がなかなか進まない要因としては、日本国内では厚生労働省が定める「ER/ES指針」や、米国ではFDAが定める「FDA 21 CFR Part11」(以降「Part11」)などのER/ES(電子記録/電子署名)規制への対応のハードルの高さが挙げられます。
従来から規制当局の監査の際に行われてきた紙の文書での提示方法から脱却ができないため、電子化を推進せずに従来どおり紙の文書での提示方法で済ませる意向が強かったからです。
加えて、医療機器メーカの業務に用いられるコンピュータシステムに必要なCSV(Computerized System Validation:コンピュータ化システムバリデーション)対応も大きな負担となっています。CSVは、一般的なシステム導入とはプロセスやテストの方法が異なっていたり、開発のレベルによってバリデーションの難易度が分かれるといったように、CSV対応によってシステム導入が複雑化かつ負担を大きくしているのです。
電子化を検討する動き
しかしながら、最近になって製品開発の電子化を検討し始める医療機器メーカが増え始めています。それには、次のような背景があると言えます。
まずは、世の中の製造業ではDXへの取り組みが加速。医療機器メーカにもその流れが押し寄せてきています。また、薬機法改正による2021年8月からの添付文書電子化の義務化のように、規制の面からもデジタル化の必要性が求められています。さらに長期に渡って刷新できずにきた基幹システムの老朽化といったことも要因となり、規制対応と業務効率化を一気に進めようとの動きが出始めているのです。
なお、FDAの「Part11」や「Part820」(QSR:品質システム規制)が遵守されていないと、当局からWarning Letterが出され、最悪のケースでは米国への出荷停止といった事態になるため、何としても確実な対応が求められるところです。
製品開発の成果物を電子化するメリット
製品開発における成果物は、Part820の設計管理においてDHF(Design History File:設計履歴ファイル)と呼ばれ、仕様書、図面、製品構成などを含む設計に関する全ての記録をデザインレビューや設計バリデーション等の各時点で保管する必要があります。これらの全てを紙にプリントアウトしキングファイルに綴じ込むといったアナログな運用では、設計変更が行われることを考えるだけでもとても非効率であり作業ミスにもつながります。これらを電子化することにより、効率的かつ確実な運用が可能になります。
但し、設計成果物はExcelやWordなどで作成される文書だけではなく、BOMや変更情報など一般的にはデータベース上のテーブルで管理されるデータも含まれます。こうした様々な成果物の運用管理にはPLM(Product Lifecycle Management)システムにより、文書以外も一元的に管理することが非常に有効です。実際に、PLMシステムでBOMなどの電子データで運用管理を実現する事例が増え始めています。
ここでは2社の事例をご紹介します。
●日機装株式会社
「Part11」に対応可能なPLMシステムの構築による医療機器ビジネスのグローバル展開を加速
 日機装様 Obbligato事例はこちら
日機装様 Obbligato事例はこちら●オリンパス株式会社
医療事業の修理業務における保守パーツ管理にPLMシステムを導入
- パーツリスト作成やマニュアルデータ管理工数を削減
- 変更通知作成や拠点への配信登録作業が約50%改善
- CSV(Computer System Validation)対応、既存ソフトウェアの回顧的バリデーション対応を実現
必要な機能をプリセットした「Obbligato医療機器テンプレート」
これらのような電子原本管理のシステム化においては、「Part11」のようなER/ES規制に対応すべく、電子署名や改ざんされていないことをトレースできる監査証跡に対応したシステムであることが非常に重要です。
さらに、BOMなどの電子データをPDF形式の電子帳票化した文書として、電子データと一緒にPLMシステム上で管理することによって、文書を中心とした成果物管理を実現する運用事例もよく見られます。その場合は、電子データと文書が一致することをシステムとして担保する必要があるため、BOMや変更情報のワークフローと連動してシステム上で自動帳票化するといった対応がよく取られています。 そこで、NECではこうした医療機器メーカにおけるPLM導入の早期実現を図るため、医療機器メーカの製品開発成果物の電子化に求められる機能群をプリセットし、「Obbligato医療機器テンプレート」として提供しています。
加えて、このテンプレートを導入する際にバリデーションへの対応による負担を軽減するため、NECではバリデーション対応を考慮したシステムの導入手法をまとめた「Obbligatoバリデーションパック」を活用することで、効率的に進めていくことができるようにしています。

医療機器テンプレートのURSをベースに導入検討が可能
PLM導入におけるバリデーションでは、PLMに対するURS(User requirements specification:ユーザ要求仕様書)を作成し、その要求が確実に満たされていることを確認することが求められます。しかし、PLMの要件を1から定義しようとすると難しい場合が少なくありません。そういった企業でも、「Obbligato医療機器テンプレート」があれば、サンプルのURSをベースに自社の強みを加味した要件を容易にまとめあげることが可能です。
さらに、サンプルのURSに記載されている要件に相当する機能を、あらかじめテンプレートとして提供していますので、できるだけ要件をサンプルのURSに寄せることで、短期間かつ高品質でのシステム導入を可能にします。
また、設計成果物は「Part820」で対応が求められる苦情やCAPA(Corrective Action and Preventive Action:是正措置・予防措置)との関連性も高く、CAPAをまわすことによって生じた設計変更でどのDHFを改版したのか、といったようにDHFとCAPAが関連性を持って運用されることが必要であり、Obbligato医療機器テンプレートの導入後、将来的に苦情やCAPAを管理する「品質イベント管理」を拡張することで、それらが一体的に運用されるシステムにステップアップすることができます。
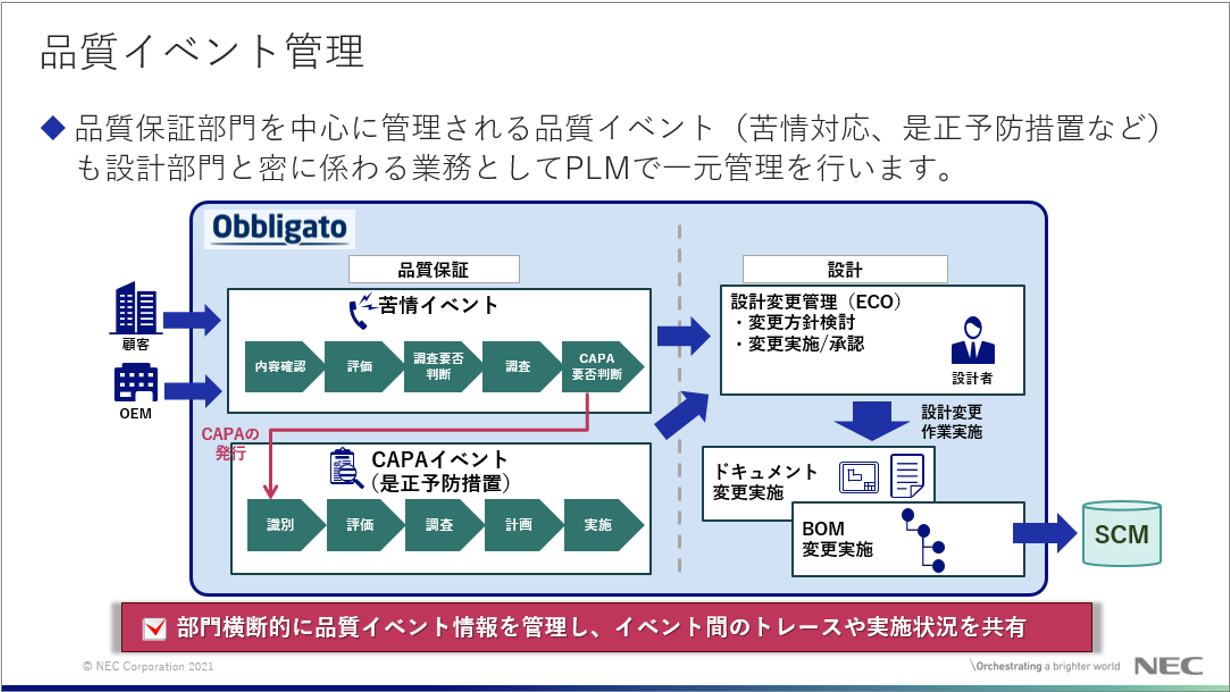
以上のように、医療機器メーカにとってER/ES規制対応やバリデーションは高いハードルとなりますが、これらを着実に実施し乗り越えて電子化を推進していく必要があります。電子化が実現できれば、規制対応とあわせて、データ共有による書類作成や問い合わせ対応といった付帯業務の削減やスピードアップなど様々な業務効率化の効果がもたらされます。さらに、製品開発QCD向上、DX推進の第一歩へと繋がります。ぜひNECと一緒に電子化を推進していきましょう。
※本サイトの法規制に関する記載内容にはNEC独自の解釈も含まれます。法規制への対応には最新情報を確認ください。
関連リンク
PLM/Obbligato
医療機器業向けソリューション
医療機器メーカに求められる『FDA 21CFR Part11、Part820』適合を推進し、技術情報や品質情報の電子化、業務効率化、品質向上を支援します。
お問い合わせ